
Recent progress in genome editing technologies, particularly with the CRISPR/Cas9 systems has opened the possibility of precise gene corrections for treating hemoglobin disorders. Our strategy is to edit the diseased human hematopoietic stem cells, as the corrected HSCs will be a renewing source of normal blood cells upon autologous bone marrow transplant. Reversing the fetal to adult hemoglobin switch is of substantial interest since persistence of high levels of HbF ameliorates clinical symptoms of SCD and β-Thalassemia patients. The increase of fetal globin could potentially lead to a valid and comprehensive strategy for the treatment of hemoglobin disorders. Clinically this is achieved through compounds like Hydroxyurea, Histone deacetylase (HDAC) inhibitors, and DNA Methyltransferase (DNMT) inhibitors, etc. Manipulation of several transcription factors like BCL11A, which acts as a critical regulator of fetal globin expression is being explored for therapeutic applications. The current focus of the Genome editing component is to develop a novel approach for the treatment of β Haemoglobinopathies. For this purpose, we plan to utilize a targeted genome engineering platform based on the CRISPR/CAS9 system to reactivate gamma-globin by gene-editing the potent gamma-globin repressor and HPFH mutations in hematopoietic stem cells (HSCs). Our approach will also be used to treat sickle cell disease by increasing the levels of functional hemoglobin and diluting the load of sickled hemoglobin levels. This approach will also benefit β-thalassemia patients by replacing defective beta globin chains of adult hemoglobin with the gamma chains.
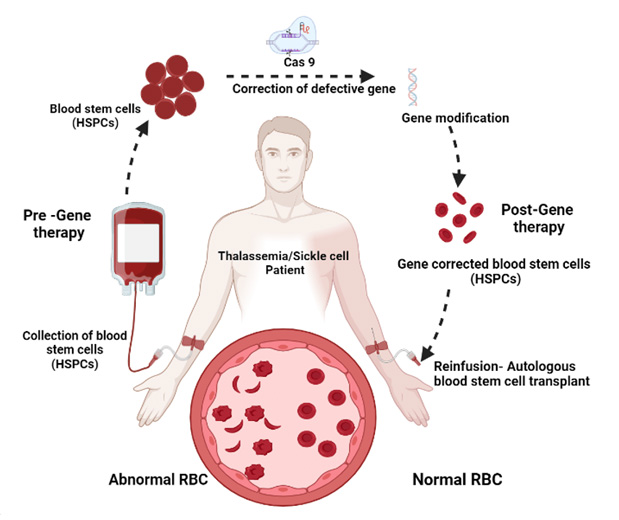
Figure: Genome editing of Hematopoietic stem cells for the treatment of beta hemoglobinopathies